Mitochondrial Hepatopathies
Dec 3, 2024
Navigate Quickly
Importance of Mitochondria
Types of Mitochondrial Hepatopathies
Primary Mitochondrial Hepatopathies
Secondary Mitochondrial Hepatopathies
Few Things to Know About Mitochondrial Hepatopathies
Laboratory Investigations
Treatment
Neonatal Liver Failure
Clinical Presentation
Laboratory Investigations
Treatment
Alpers Syndrome
Clinical Presentation
Investigations
Pearson Marrow Pancreas Syndrome
Clinical Presentation
Important Questions

Out of all the mitochondrial diseases, liver involvement is 20%. It is the relatively more common disorder after CNS and neuromuscular diseases in infants.
Importance of Mitochondria
It is the powerhouse of a cell. It plays an important role in ATP formation. Electron transport chain (ETC) enzymes are present in mitochondria. Hence it is involved in energy production. Any injury to mitochondria can release pro-apoprotein triggers into the cytosol and produce cell apoptosis. cyt C. The liver, being involved in many metabolic processes, is also involved in various mitochondrial diseases.
Types of Mitochondrial Hepatopathies
There are mitochondrial hepatopathies. In hepatopathies, the primary defect is found in the primary defect mitochondria. Either deficiency of any mitochondrial enzyme or any function is affected.
In hepatopathies secondary, defects lie elsewhere. A drug or a secondary insult might be the underlying cause, affecting mitochondria.
Also read: Neonatal Respiratory Monitoring
Primary Mitochondrial Hepatopathies
Primary mitochondria are of the following types based on their causes:
- Electron transport chain/respiratory chain:
- Neonatal liver failure
- Mitochondrial DNA Depletion Syndrome
- Alper's syndrome
- Pearson's Marrow Pancreas Syndrome
- Navajo Neurohepatopathy
- Villous Atrophy Syndrome
- GRACILE Syndrome
- Fatty Acid Oxidation Defects
- Urea Cycle Enzyme Deficiencies
- PEP Carboxykinase Deficiency
- Electron Transfer Flavoprotein And Electron Transfer Flavoprotein Dehydrogenase Deficiencies
Secondary Mitochondrial Hepatopathies
Secondary mitochondrial hepatopathies are seen in conditions like Reye's syndrome. Copper and Iron Overload Syndromes: Wilson's disease is an example of copper overload disease, and various types of hemochromatosis are examples of iron overload disease. Drug-induced: Valproate and similar anti-epileptic drugs are found to trigger the condition. Toxin-induced: This includes carbon tetrachloride in various types of industrial reagents. And Cholestasis. Alpha-1 Antitrypsin Deficiency
Also read: Autoimmune Hepatitis: Types, Clinical Presentation, Diagnosis And

Few Things to Know About Mitochondrial Hepatopathies
These diseases show clinical presentation. The variable reason for this can be explained as follows:.
Mitochondria usually shows maternal inheritance, indicating every individual receives mitochondria from their mother and none from their father. The division of mitochondria occurs as a random segregation and not as an exact. As a result, if a mother has defective mitochondria, then the defective mitochondria inherited by her different children would also differ.
A single individual or a single cell would inherit different types of mitochondria. Normal and abnormal mitochondria may be present in different cell lineages. This phenomenon of heterogeneous inheritance of mitochondria is referred to as heteroplasmy. Another reason for variable clinical presentation is that mitochondria are highly prone to mutation.
Hepatic features range from
- Simple hepatic steatosis
- Hepatomegaly
- Acute hepatic failure
- Hronic liver disease
- Cirrhosis
- Failure to thrive
- Vomiting
- Multisystem involvement
which cannot be easily explained, should raise suspicion.
Also read: Alagille Syndrome – Clinical Features And Diagnosis
Laboratory Investigations
As a screening test, the plasma lactate to pyruvate ratio levels is tested. The lactate:pyruvate ratio being more than or equal to is a positive indicator of the disease. 25 or 25:1. Since failure to thrive and anemia can manifest in several mitochondrial diseases, blood ammonia levels and CBC or complete blood counts are While checking blood ketone levels, quantitative measurements of 3-hydroxybutyrate and aceto acetate levels are also checked.
Checking the level of plasma acylcarnitine is beneficial in cases of defective metabolic conditions or disorders like fatty acid oxidation defects. Urine GCMS is tested for organic acids. ABG and blood pH levels are tested to look for metabolic acidosis. If neurological involvement is suspected, then CSF is also tested. After all these, a liver biopsy is sometimes done as a second line of investigation.
Treatment
The treatment of mitochondrial hepatopathies is mainly supportive. If the patient has acidosis, then periodically, sodium bicarbonate is given. If the patient has anemia or thrombocytopenia, then a transfusion is given. Additionally, nutritional support is given. Commonly, cocktail of multivitamins and antioxidants is given. This cocktail contains CoQ10, vitamin B complex, vitamins C and E, glutathione derivatives, and sometimes lycopene. For some patients, oral L-carnitine is recommended to be given daily. But the above-said cocktail and L-carnitine have not been scientifically proven to alter the course of the disease. The final method of treatment is liver transplantation. The orthotopic liver transplants performed are found to be useful in secondary hepatopathies. (The same has been said to be of little to no use if there has been any significant CNS involvement).
Also read: Abdominal Tuberculosis in Children: Symptoms, Diagnosis
Neonatal Liver Failure
This condition mainly arises due to defective electron transport chain (ETC) complexes. There would be either a deficiency or complete absence of ETC complex enzymes. The cytochrome-C oxidase, or Type IV complex deficiency, is the most common cause of neonatal liver failure compared to Type III and Type I complex deficiencies. The mutations that happen here are usually spontaneous and not due to any genetic reasons.
Clinical Presentation
The clinical onset of this condition occurs within the first 6 weeks of life. The clinical manifestations are seen as 3 types of features. Those are :
- General features
- Acute hepatic failure features
- CNS involvement features.
The general features are failure to thrive, poor weight gain, lethargy, and irritability by the child. The features of acute hepatic failure exhibited by the child include coagulopathy, hypoglycemia, ascites, and altered sensorium. The features related to CNS involvement shown by the child are focal seizures (can have secondary generalizations and multiple types), apneic episodes, and depressed sensorium.
Laboratory Investigations
The main 2 investigations were done to assess neonatal liver failure.
The first is the screening test, or the plasma lactate to pyruvate ratio. This ratio would be more than or equal to 25. The other one is the evaluation of plasma 3-hydroxybutyrate to acetoacetate ratio, which would be more than or equal to 4. Both of these values are strongly suggestive of neonatal liver failure.
Treatment
The treatment therapies done for neonatal liver failure are supportive measures. More than 95% of neonates die within 2 weeks, indicating the poor outcome of the therapy
Also read: Gastrointestinal Foreign Bodies in Children: Bezoars & Ingestion
Alpers Syndrome
This disease is also called Alpers-Huttenlocher Syndrome or Alpers Hepatopathic Poliodystrophy.
The mode of inheritance is autosomal recessive.
Mutations in the code for the catalytic subunit POLG gene of mtDNA. (The POLG gene is present on chromosome number 15). Some patients also have ETC Complex I deficiency. This disease is an example of somatic mutation causing abnormalities in the mitochondrial pathway. The clinical representation is similar to acute liver failure in newborns, and the only main difference is the CNS features are more predominant and more of a sedate course. There is no specific treatment for this disease. While neonates of acute liver failure die within 2-6 weeks, patients of Alper's disease tend to survive up to 10–12 weeks.
Clinical Presentation
The neurological features presented are poor feeding, hypotonia, partial seizures (which could later turn into Status epilepticus), neuroregression, cerebellar ataxia, etc. The neuro-regression shown here is episodic, irregular, and often aggravated or precipitated by intercurrent disease. The hepatic features are expressed as hepatomegaly with elevated bilirubin levels or acute liver failure (fulminant outcome).
Investigations
Raised lactate level in blood and CSF, along with plasma lactate to pyruvate ratio over 25. EEG shows the high amplitude and slow activity with polyspikes. Atrophy and abnormalities in the occipital and temporal lobes are found in neuroimaging. As there are no specific treatment modalities for this condition, death invariably occurs between the ages of 2 and 12 years.
Also read: Neonatal Cholestasis: Causes, Diagnosis, and Treatment in Infants
Pearson Marrow Pancreas Syndrome
The genetic basis of this condition is mtDNA deletion, which results in deficiencies in ETC Complexes I and III. The onset of the condition occurs at the neonatal stage or infancy.
Clinical Presentation
Severe macrocytic anemia, variable neutropenia, and thrombocytopenia are seen in blood. On bone marrow examination, ringed sideroblasts are found. Pancreatic fibrosis will also occur due to diarrhea and fat malabsorption. Partial villous atrophy of the small intestine also occurs. The liver exhibits features like hepatomegaly, steatosis, and cirrhosis upon endoscopy. This condition is biomarker 3-Methyl-Glutaconic Aciduria.
Important Questions
Q. A patient with hepatomegaly presented to the emergency department with status epilepticus. The patient was started on a benzodiazepine. Since there was no response to seizures, valproate was started. Starting valproate began to worsen the condition as the patient began to show features like raised intracranial pressure, worsening of the patient, and alleviation in the hepatic enzymes. Which disease would you suspect here?
Ans. Alpers Disease
Q. Which of the following should be avoided in a child suspected of having a hepatic mitochondrial disorder?
A. Normal Saline
B. Ringer's lactate
C. 5% Dextrose
D. Packed Red blood cell transfusion
Ans. Ringer's lactate. It should be avoided because patients with liver dysfunction may be unable to metabolize lactate.
Q. A 4-month-old child is found to have pallor, petechiae, and steatorrhea. A peripheral blood smear revealed macrocytic anemia. USG revealed fatty liver, and BM findings are as shown. What is the likely diagnosis?
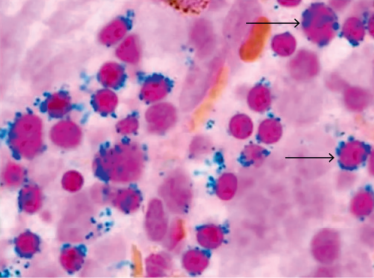
Ans. Pearson Marrow Pancreas Syndrome
Also read: NEET SS Preparation Strategy No One Talks About!
Hope you found this blog helpful for your NEET SS Pediatrics Gastroenterology Preparation. For more informative and interesting posts like these, keep reading PrepLadder’s blogs.

PrepLadder Medical
Get access to all the essential resources required to ace your medical exam Preparation. Stay updated with the latest news and developments in the medical exam, improve your Medical Exam preparation, and turn your dreams into a reality!
Top searching words
The most popular search terms used by aspirants
- NEET SS Pediatrics Gastroenterology
- NEET SS Pediatrics Gastroenterology and Hepatobiliary Preparation
PrepLadder 4.0 for NEET SS
Avail 24-Hr Free Trial